
Soluções:
#1
Considerando a molaridade igual a um por se tratar de condições padrões. Usando a estequiometria da questão chegamos que

Mas note que nosso  é muito pequeno, logo
é muito pequeno, logo  assim podemos desprezar
assim podemos desprezar  .
.

#2
Ocorrerá a seguinte reação de decomposição:

Como o pentóxido de dinitrogênio é o único reagente e a reação é de ordem 1, temos que sua lei de velocidade será:
![v = k[N_{2}O_{5}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_01ef54ef9986043712a4a4fef90d77ff.gif?ssl=1)
A massa molar do pentóxido de dinitrogênio é:

De modo que haverá:

Resultando em:
![[N_{2}O_{5}] = \frac{3,194 \cdot 10^-2}{0.750} = 4,258 \cdot 10^{-2} mol/L](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_9cb1759362c760dbdb471a86ac6d7994.gif?ssl=1)
Logo a velocidade inicial pedida será:

#3
Como sabemos,  é a razão entre as atividades dos produtos elevadas aos coeficientes pelas atividades dos reagentes elevadas aos seus coeficientes, também sabemos que as atividades de um sólido e de um líquido são ambas iguais a 1 e que a atividade de um gás é sua pressão parcial e a de um soluto é sua concentração.
é a razão entre as atividades dos produtos elevadas aos coeficientes pelas atividades dos reagentes elevadas aos seus coeficientes, também sabemos que as atividades de um sólido e de um líquido são ambas iguais a 1 e que a atividade de um gás é sua pressão parcial e a de um soluto é sua concentração.
a) 
b) ![Q = [H_{3}PO_{4}]^4 \cdot [H_{2}S]^{10}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_8672699b9fa0c9416be29b5ff9c92f31.gif?ssl=1)
c) 
#4
emos as seguintes semi-reações:


Assim  . Usando a equação de Nernst conseguimos o resultado desejado:
. Usando a equação de Nernst conseguimos o resultado desejado:

Substituindo:

#5
Considerando as cinco substâncias, temos:





Soluções que apresentam maior concentração de partículas possuem menor valor de temperatura de congelação. Pelas dissociações acima, temos: I V II III IV
#6
Devemos primeiro calcular os valores dos Q's com todos os íons dissolvidos.

Podemos ver que ambos os Q's são maiores que seus respectivos k's. Dessa forma podemos considerar as concentrações dos íons como sendo a necessária para alcançar os k's e manter as proporções estequioétricas.
![K_{BaF_{2}} = [Ba^{2+}][F^-]^2 \Rightarrow 1,0 \cdot 10^{-6} = x\cdot (2x)^2 \Rightarrow](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5c09b20cc28f72804f4fa51966330756.gif?ssl=1)
![\Rightarrow [Ba^{2+}] = 0,0063 Mol/L \ \text{ e } \ [F^-] = 0,0126 Mol/L](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_20419788464053d4ca6e73331329f653.gif?ssl=1)
![K_{Pb(OH)_{2}} = [Pb^{2+}][OH^-]^2 \Rightarrow 1,2 \cdot 10^{-15} = x\cdot (2x)^2 \Rightarrow](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_7a13b56b49f018a0ced0e4a84d58ada0.gif?ssl=1)
![\Rightarrow [Pb^{2+}] = 6.7 \cdot 10^{-6} M \ \text{ e } \ [F^-] = 1.34 \cdot 10^{-5} M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_9bf9dde066d0fcda4241d8ca18973087.gif?ssl=1)
Assim obtivemos os valores desejados.
#7
Sabemos que a massa molar de nosso composto é:  . Agora, basta sabermos a molaridade do
. Agora, basta sabermos a molaridade do  dissolvido:
dissolvido:

 e dois mols de
e dois mols de  . Substituindo na equação do
. Substituindo na equação do  :
: ![K_{ps} = [Pb^{2+}][Cl^-]^2 \Rightarrow K_{ps} = [0.0159][0.0318]^2 = 1,61 \cdot 10^{-5}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_6194c9ebe3b7721ad66580320452c7c3.gif?ssl=1)
#8
a) Temos que 
b)Novamente como temos um ciclo de carnot: 
c) Podemos notar que a eficiência não é diretamente proporcional à eficiência e que, dada uma diferença de temperatura, é mais vantajoso termos sempre a menor temperatura mínima.
#9


Por outro lado, a molalidade da solução é  , onde m é a massa molar do soluto.
, onde m é a massa molar do soluto.

O álcool correspondente a esta massa molar é o etanol,  .
.
#10
Pela Lei de Henry, temos:

Portanto, 1,0 litro d'água dissolve  mol de
mol de  .
.
Como estamos na STP o volume molar é 24,7 L. Desse modo:

#11
Como a solução é suficientemente diluída:
( [H+]água + [H+]ácido ) . [OH-] = 10-14
A única fonte de OH- é a autoionização da água. Assim:
( [H+]água + [H+]ácido ) . [H+]água = 10-14
( [H+]água + 10-8 ) . [H+]água = 10-14
( [H+]água)2 + 10-8 . [H+]água - 10-14 = 0
Encontramos [H+]água = 9,51 . 10-8 mol/L
Logo, [H+]total = 9,51 . 10-8 + 1.10-8
[H+]total = 1,05 . 10-7 mol/L => -log [H+]total = pH = 6,978
#12
Equações:
[HClO] + [ClO-] = 1.10-8 (balanço molar)
[H+] = [OH-] + [ClO-] (balanço de carga)
[H+].[OH-] = 10-14 (constante de autoionização da água)
Ka = ([ClO-][H+]) / [HClO] (constante de acidez do HClO)
Fazendo as devidas substituições, temos:
Ka = ([ClO-][H+]) / [HClO]
10-7,497 = ([ClO-][H+]) / (1.10-8 - [ClO-])
3,18.10-8 = (([H+] - Kw/[H+]) . [H+])) / (1.10-8 - ([H+] - Kw/[H+]))
3,18.10-8 = ([H+]2 - Kw) / (1.10-8 - [H+] + (Kw/[H+]))
3,18.10-16 - 3,18.10-8 . [H+] + ((3,18.10-22)/[H+]) = [H+]2 - 10-14
Rearranjando, obtemos:
[H+]3 + 3,18.10-8 . [H+]2 - 1,032.10-14 . [H+] - 3,18.10-22 = 0
A única solução positiva encontrada é [H+] = 1,0121 . 10-7
-log[H+] = pH = 6,995
#13
Verdadeira. A reação de formação do dímero é exotérmica, pois ocorre formação de ligação química. Isso também pode ser verificado através do deslocamento do equilíbrio. Quando se diminui a temperatura, pelo Princípio de Le Chatelier, o sentido reacional exotérmico é favorecido. Logo, como o gás no recipiente fica incolor, percebemos que a reação de formação de  é favorecida. Consequentemente, ela é exotérmica.
é favorecida. Consequentemente, ela é exotérmica.
Verdadeira. Com o aumento de pressão, o equílibrio se desloca de forma que os gases ocupem o menor volume possível, implicando diminuição do número de mols da mistura. Assim, a formação de é favorecida. Esse fato é evidenciado pelo desaparecimento da coloração castanha.
Verdadeira. Quando o sistema absorve calor, o equilíbrio é deslocado no sentido reacional exotérmico, favorecendo a dissociação do dímero e o aumento da concentração de  .
.
#14
a) ∆Tc = 1,86. (0,8 / 10) . (1 / 4,0.105)
∆Tc = 3,72.10-7 oC
п = (0,8.10-3g / 4,0.10-5 g/mol).(1 / 10.10-3L).(0,082 atm.L / mol.K). 298,15K . (760 torr/ 1 atm)
п = 3,716.10-3 torr
b) п = (0,1 + 3,716.10-3) torr . (1 atm / 760 torr)
п = (0,8.10-3 / M).(1 / 10.10-3L).(0,082 atm.L / mol.K). 298,15K . (760 torr/ 1 atm)
M = 1,43.104 g/mol
#15
a) Comparando os valores de Kps, deduz-se que o  é o hidróxido menos solúvel. Dessa forma:
é o hidróxido menos solúvel. Dessa forma:
![1.10^{-3} \cdot [OH-]^2 = 1,6.10^{-19}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_c1291d5c838022cc2c7f5eb9b1fc5fea.gif?ssl=1)
![[OH-] = 5,43.10^{-6} mol/L](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_a3db6311058580eb3ec7ebbe8a9185a9.gif?ssl=1)

Uma vez que é o hidróxido de menor solubilidade, os outros compostos requerem pHs ainda maiores (maior concentração de OH-) para que ocorra precipitação. Assim, é necessária uma solução básica.
b) Mediante o controle de pH, é possível separar os cátions por precipitação seletiva.
Separando o  :
:
![1.10^{-3} \cdot [OH-]^2 = 1,3 \cdot 10^{-6}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_60f041ecf9c5edf68549829cc0c7aa2b.gif?ssl=1)
![[OH-] = 0,036 mol/L](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_d6316df1337cff8a09242c60e34abc38.gif?ssl=1)

Separando o  :
:
![1.10^{-3} . [OH-]^2 = 5.10^{-3}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_a989181409674cd959c9fed3978c4a9c.gif?ssl=1)
![[OH-] - 2,24 mol/L](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_4dfd06cdfd397401b005517f4889367a.gif?ssl=1)
 ]
]
#16
a) ![Kb = [pyH^+][OH^-]/[py]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_7460e746b8dcd587c7b6ffbbd95dee27.gif?ssl=1)

![x = [OH^-] = 2,07.10^{-5} M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5e115e6cbd46c38efa813450e6cd6b0d.gif?ssl=1)

b) i) Após a adição de  de
de  , sobram
, sobram  de piridina e são produzidos
de piridina e são produzidos  de piridínio.
de piridínio.
![pOH = pKb + log ([pyH^+]/[py])](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_49d25841ea63940db82ec82e41bd55ee.gif?ssl=1)


ii) No ponto de equivalência, temos ![[C_5H_5NH^+] = 0,08M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_e4fecc5fe513a86fe40feb9b28ddc7bf.gif?ssl=1) . O piridínio hidrolisa formando piridina e íons
. O piridínio hidrolisa formando piridina e íons  .
.
![Kb = [pyH^+] \cdot Kw / [H+][py]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_a88ccaaa3bc59d0ec0fb98b7ad50a905.gif?ssl=1)
![Kb = [pyH^+] \cdot Kw / [H+]^2](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_fab2833fd389eba4a188d77b05816065.gif?ssl=1)
![[H^+] = 6,70.10^{-4} M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_07563caf6bb1f40924e188e86933522c.gif?ssl=1)

![c \cdot Kps = [Mg_2^+][OH^-]^2](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5649a71527555f8aa7e5caccb7c6fc54.gif?ssl=1)
![[Mg_2^+] = 5,6.10^{-12} / (2,07.10^{-5})^2](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_27cfabf2ae225d11d9e077afc985e99d.gif?ssl=1)
![[Mg_2^+] 1,3.10^{-2} M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_672a04a7410ca9095d636331f8175470.gif?ssl=1)
d. A concentração máxima de hidróxido é dada por
![[OH-] = \sqrt{Kps/[Mg_2^+]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_feee0adaece2c376b047b6f653dbaf85.gif?ssl=1)
![[OH-] = 7,48.10^{-6} M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5143c8b2668da90c900b8ed0faed83cb.gif?ssl=1)
Substituindo:
![pOH = pKb + log([pyH+]/[py])](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_230643235ab0ca4cf3db2bc6ca586555.gif?ssl=1)
![5,13 = 8,75 + 0,624 + log [pyH^+]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_3e44c32b5bfd0501ecfa39454d7dabbf.gif?ssl=1)
![[pyH^+] = 5,7.10^{-5} M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_a527aa37eb68dbe25570ad8cf597387f.gif?ssl=1)
#17
A espontaneidade de um processo pode ser avaliada por

Onde  é necessariamente negativo para um processo espontâneo. Assim, tratando-se de um processo endotérmico e espontâneo, deve ser satisfeita a condição
é necessariamente negativo para um processo espontâneo. Assim, tratando-se de um processo endotérmico e espontâneo, deve ser satisfeita a condição  para que
para que .
.
#18
 G's individuais.
G's individuais. +
+ 

 +
+  G =
G =
 =
=  +
+ 

b) ![[Pb^{2+}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5ce476075e749cfcfe5d192b128a90a0.gif?ssl=1) = 3,4.
= 3,4. M
M
c) O potencial pode ser calculado a partir das duas semi-reações:
i) + 2e- Pb +
 - (0,0592/2) log []
- (0,0592/2) log []
 = -0,394 - 0,0296 log [0,025]
= -0,394 - 0,0296 log [0,025]

 +
+  Pb - (0,0592/2) . log [] = -0,126 - 0,0296 log [3,4.]
Pb - (0,0592/2) . log [] = -0,126 - 0,0296 log [3,4.]#19

 =
= 

![(\frac{[H^+][OH^-]}{1.10^{-14}})](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_7890bee84bbd924f2a79b946884916ee.gif?ssl=1) =
= 

Na água pura, ![[H^+] = [OH^-]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_6bba4f150eebd132dd299f44ece24d5d.gif?ssl=1)
![[H^+]^2 = -31,84](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_439864ed07be93ecd2dd44ee31a9f5ec.gif?ssl=1)
![\Rightarrow [H^+] = 1,22\times 10^{-7} \frac{mol}{L}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_bb25be7649562a31fdb3739d11b1a7ea.gif?ssl=1)
![-log[H^+] = pH = 6,91](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_c2deee2936634bbc8c78213f31767fd7.gif?ssl=1)
#20
A solubilidade molar do iodato de tório(IV) pode ser escrita como
![[Th^{4+}][IO_3^-]^4 = K_{ps} \Rightarrow S \cdot (4S)^4 =K_{ps}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_1b1a3d6385709a3cae19e372d6a37263.gif?ssl=1)

#21










#22
a) As equações balanceadas são:


b) A partir de :



De e :



![[Cu^{2+}] = \frac{2,31 \cdot 10^{-4} mol}{0,02000 dm^3} = 1,15 \cdot 10^{-2}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_2a8e87161b3a28a6279d7e57c4ccf7f4.gif?ssl=1)

![\Rightarrow [IO_3^-] = 2 [Cu^{2+}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_3ebc1f16c2261ad854cff005d5245558.gif?ssl=1)
![K_{ps} = [Cu^{2+}][IO_3^-]^2 = 4 \cdot [Cu^{2+}]^3 = 4 \cdot (1,15 \cdot 10^{-2})^3 = 6,08 \cdot 10^{-6}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_66250ac97e72c05d2da4aea1aad3ae8c.gif?ssl=1)
#23
a) ![K_1 = \frac{[H^+][CH_3COO^-]}{[CH_3COOH]} = \frac {(\alpha _1 + \alpha _2)c \cdot \alpha _1 c}{(1 - \alpha _1)c} = \frac {(\alpha _1 + \alpha _2) \cdot \alpha _1 c}{(1 - \alpha _1)}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_dd6ee7c1202505d5cbde71d372f427cd.gif?ssl=1)
![K_2 = \frac{[H^+][ClO^-]}{[HClO]} = \frac{(\alpha _1 + \alpha _2)\alpha _1 c}{1 - \alpha _2}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_c04a0fe258ed9769e55720e990cabc77.gif?ssl=1)
Como  , consequentemente
, consequentemente  . Assim, é válida a aproximação
. Assim, é válida a aproximação  .
.


Encontramos  .
.
Dividindo por :

Substituindo  , encontramos
, encontramos  .
.
b)  , sendo que
, sendo que  .
.
 .
.
c) ![[H^+] = \alpha _1 c + \alpha _2 c = (0,125 + 2,94 \cdot 10^{-4}) \cdot 10^{-3} \simeq 1,25 \cdot 10^{-4}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_f226edfc64fbf72c7612555e381cc745.gif?ssl=1)


#24
Primeiramente, leve em conta que, em solução aquosa de  , a solubilidade de sais de prata aumenta consideravelmente, devido à formação do complexo
, a solubilidade de sais de prata aumenta consideravelmente, devido à formação do complexo  .
.








Visto isso, sabemos que maior solubilidade relaciona-se ao sal em que a ligação  é mais fraca. No
é mais fraca. No  , a ligação apresenta maior caráter iônico que em
, a ligação apresenta maior caráter iônico que em  , devido à menor polarizabilidade do íon
, devido à menor polarizabilidade do íon  . De fato, a ligação com maior caráter iônico (e consequentemente menor caráter covalente) apresenta-se mais fraca, quebrando-se mais facilmente, o que explica a maior solubilidade do .
. De fato, a ligação com maior caráter iônico (e consequentemente menor caráter covalente) apresenta-se mais fraca, quebrando-se mais facilmente, o que explica a maior solubilidade do .
#25
O congelamento abaixo da temperatura normal remete à propriedade coligativa de crioscopia. Como todas as outras propriedades coligativas, ela é unicamente influenciada pela quantidade de partículas em questão. Assim, embora as soluções de cloreto de sódio e glicose possuam a mesma concentração molar, a de cloreto de sódio possui maior quantidade de partículas em solução, pois o sal dissocia-se em  e , apresentando maior abaixamento da temperatura de fusão.
e , apresentando maior abaixamento da temperatura de fusão.
#26
A constante de equilíbrio fornecida representa a seguinte reação:





Pelo Princípio de Le Chatelier, a adição de  ao sistema desloca o equilíbrio para o lado dos reagentes, de forma a consumir o excesso de produto. Isso também pode ser verificado pela expressão de :
ao sistema desloca o equilíbrio para o lado dos reagentes, de forma a consumir o excesso de produto. Isso também pode ser verificado pela expressão de :

![\frac {[CO] \cdot [H_2]^3}{[CH_4] \cdot [H_2O]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_8e5088f54f61bee0ddbb4f3e12310719.gif?ssl=1) . Para que se mantenha constante, deve haver aumento na concentração dos reagentes (denominador da fração).
. Para que se mantenha constante, deve haver aumento na concentração dos reagentes (denominador da fração).
#27
A reação apresenta trata da cisão homolítica de moléculas de  . Sabemos que a dissociação dessa molécula diatômica compreende a quebra de uma ligação estável, formando dois radicais
. Sabemos que a dissociação dessa molécula diatômica compreende a quebra de uma ligação estável, formando dois radicais  espécies altamente reativas , o que é desfavorável energeticamente.
espécies altamente reativas , o que é desfavorável energeticamente.
Assim, a reação apresenta  . Extraída essa informação, podemos solucionar o problema.
. Extraída essa informação, podemos solucionar o problema.


 Um aumento da temperatura desloca o equilíbrio para a direita, levando a uma maior produção de
Um aumento da temperatura desloca o equilíbrio para a direita, levando a uma maior produção de  , pois há maior quantidade de energia disponível para a quebra da ligação
, pois há maior quantidade de energia disponível para a quebra da ligação  .
.
 Quando o volume do sistema é diminuído, o equilíbrio se desloca para o lado com menor número de mols de gás (menor volume ocupado). Logo, o sentido inverso é favorecido.
Quando o volume do sistema é diminuído, o equilíbrio se desloca para o lado com menor número de mols de gás (menor volume ocupado). Logo, o sentido inverso é favorecido.
 A adição de xenônio não desloca o equilíbrio. Embora aumente a pressão total do sistema, reduz proporcionalmente as pressões parciais dos gases, anulando quaisquer efeitos sobre o equílibrio.
A adição de xenônio não desloca o equilíbrio. Embora aumente a pressão total do sistema, reduz proporcionalmente as pressões parciais dos gases, anulando quaisquer efeitos sobre o equílibrio.
#28
O equilíbrio de dissociação proposto é 
Antes de ser atingido o equilíbrio, temos 
 de
de  e
e  de
de 
Para atingir-se o equilíbrio, são consumidos ![\alpha \cdot [A_2B_4] = \alpha \cdot 1 = \alpha](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_a7245241e652103d71fd9b34ec346e0e.gif?ssl=1) de e são produzidos
de e são produzidos 

 de .
de .
Assim, a constante de equilíbrio  pode ser escrita como
pode ser escrita como  . Como temos o valor de
. Como temos o valor de  , calculamos a quantidade de dissociada
, calculamos a quantidade de dissociada  .
.


 %
%
#29
a) Se o reator tende a esquentar conforme a reação ocorre, então a reação é claramente exotérmica. Note-se também que a reação ocorre com diminuição da quantidade de reagentes gasosos. Assim, a maior conversão (isto é, situação em que o equilíbrio está mais deslocado para os produtos) se dá em baixas temperaturas (segundo Le Chatelier, aquecer reações exotérmicas desloca o equilíbrio para os reagentes) e em altas pressões (segundo Le Chatelier, reações em que Δngás<0 deslocam para produtos com o aumento da pressão).
b) Sim, faz sentido: segundo o princípio de Le Chatelier, altas pressões favorecem a formação de produto (nesse caso, amônia); assim, usar pressão elevada (como 100 atm) aumenta a conversão.
c) Do ponto de vista termodinâmico, não faz sentido: altas temperaturas favorecem a reação inversa (decomposição da amônia em seus elementos), o que diminui a conversão. A razão industrial para o uso de altas temperaturas é cinética: acelerar a formação de amônia a partir de seus reagentes, uma vez que mesmo o processo catalisado é considerado lento demais para as atividades industriais (isto é, sacrifica-se uma maior conversão para que a produção seja mais rápida).
#30
I) Falsa: mesmo que não consumidos no processo global, catalisadores são espécies que interagem com os reagentes (ou intermediários formados) e depois são regenerados em passo posterior – é impossível que uma espécie altere a reação sem que participe da mesma de alguma forma.
II) Verdadeira.
III) Falsa: na catálise homogênea, catalisador e reagente obrigatoriamente encontram-se na mesma fase; do contrário, trata-se de catálise heterogênea.
IV) Falsa: catalisadores não alteram constante de equilíbrio, uma vez que não alteram os níveis energéticos nem de reagentes nem de produtos, pois não se mantêm ligados a nenhum deles. Logo, não se pode dizer que o ácido aumenta a extensão da reação.
V) Verdadeira.
VI) Verdadeira.
#31
a) 6 CO2 (g) + 6 H2O(v) → C6H12O6 (aq) + 6 O2 (g)
b) Diminuição de entropia, uma vez que há uma diminuição da quantidade de compostos gasosos de 12 para 6 mol por mol de glicose gerado (note que as moléculas pequenas de reagente se combinam formando uma molécula de glicose de alta massa molecular).
c) Endotérmico: perceba que a fotossíntese é o oposto do processo de combustão completa da glicose; uma vez que combustões são invariavelmente exotérmicas, tem-se por consequência que a fotossíntese é um processo endotérmico.
d) Sabe-se que a energia livre de Gibbs, ΔG, é dada por:
ΔG = ΔH – TΔS
Como ΔH>0 e ΔS<0, conclui-se que ΔG>0 em qualquer temperatura, isto é, a fotossíntese em si é um processo endergônico e consequentemente não espontâneo (desfavorável) termodinamicamente em qualquer temperatura.
e) À primeira vista, parece contra intuitivo que a fotossíntese ocorra, uma vez que é um processo endergônico a qualquer temperatura, que é o que indica a resposta do item d. De fato, esse processo isolado é desfavorecido termodinamicamente e não deveria acontecer. O que explica a ocorrência desse fenômeno é o fato de que ocorrem reações acopladas a ele que fornecem a energia necessária para que esse processo seja possível (a necessidade de luz para a ocorrência do processo está relacionada de certa forma a isso).
#32
a) Como 12 dos 119 aminoácidos são glicinas, há apenas 117 centros quirais na lisozima - cada apresenta configuração R ou S (isto é, duas possibilidades). A enzima funcional apresenta uma configuração definida para cada um desses centros, logo, há apenas uma combinação desses 117 centros que é funcional. Como há duas possibilidades para cada centro, há no total 2117 possibilidades, uma das quais é a desejada. Logo, o rendimento é 2-117=6,02.10-34%
b) Para obter 120mg de enzima funcional:
m = 120.10-6kg / 6,02.10-36= 1,99. 1031kg = 3,34.106 Terras
c) I) Falsa. A dependência da constante de velocidade (e consequentemente da velocidade) com a energia de ativação é exponencial, como expresso pela Lei de Arrhenius, não linear, como proposto pela alternativa.
II) Verdadeira. Como o processo é de primeira-ordem, depende apenas da constante de velocidade de forma inversamente proporcional: se a constante aumenta 10000 vezes, o tempo de meia-vida cai de 10000 vezes.
III) Verdadeira. Essa relação, por mais que uma aproximação, é razoavelmente válida para prever alterações na velocidade de reação (cada 10°C de aquecimento dobraria aproximadamente a velocidade, segundo essa relação).
IV) Falsa. Como a reação é catalisada, há um ponto em que haverá a saturação, isto é, todas as moléculas da enzima se encontrarão ligadas a moléculas de substrato, de modo que deixa de importar a concentração de substrato, uma vez que o fator limitante passa a ser a quantidade de enzima. Assim, o aumento de substrato leva a aumento da velocidade até a saturação, em que se passa a observar uma velocidade máxima.
#33
a) Como busca-se a energia por mol de hidrogênio, escreva-se a reação com coeficiente 1 para esse reagente:
H2 + ½ O2 → H2O
Quando se trabalha com energias de ligação, tem-se que:
ΔH = E(queb) – E(form) = {(H-H) + ½ (O=O)} - 2.(O-H) = {436 + 495/2} - 2.463 = -242,5kJ mol-1
b) Pela equação de Clapeyron:
PV = nRT = mRT/M
m = MPV/RT = 2.0,25.100 000.103/250.0,082 = 2,44.106 g H2 = 2,44t H2
c) O número de mols de H2 presente é de 2,44.106/2 = 1,22.106 mol. Do valor de ΔH do item a:
Q = n.ΔH = 1,22.106 . (-242,5.103) = 2,96.1011J = 296 GJ
d) Da definição de potência:
Pot = Q/Δt = 2,96.1011/ 30 = 9,86.109 W = 9,86 GW
#34
a) Uma reação é elementar se ocorre em uma única etapa, conforme representa diretamente sua equação química (a reação ocorre em uma única colisão, de forma direta, sem que haja vários passos ou reações diferentes).
b) A lei de velocidade esperada é da forma v = k[A][B]2[C]7, nessa situação.
c) Não, é improvável que essa reação ocorra de forma elementar: se o fosse, necessitaria da colisão simultânea de 10 moléculas com geometria e energia adequadas, o que é uma situação altamente improvável no contexto do movimento caótico das partículas proposto pela Teoria das Colisões – de modo que seria um processo extremamente lento, o que contraria as observações experimentais apresentadas.
#35
a) A técnica para obter os coeficientes é dividir leis de velocidades de diferentes experimentos, considerando que a constante de velocidade é igual para todos:
Utilizando os experimentos II e III, obtém-se o coeficiente a do reagente A:
(0,230/0,115)a = 0,1370/0,0484 ⇔ a = [log 0,1370/0,0484]/[log 0,230/0,115] = 1,50
Utilizando os experimentos III e IV, obtém-se o coeficiente b do reagente B:
(0,175/0,230)1,50. (0,050/0,030)b = 0,1174/0,1370 ⇔ b = 0,50
Utilizando os experimentos I e IV, obtém-se o coeficiente c do reagente C:
(0,175/0,050)1,50 . (0,050/0,030)c = 0,1174/0,0158 ⇔ c = 0,25
A lei de velocidade experimental é v = k[A]1,50[B]0,50[C]0,25.
Conclui-se que a reação não pode ser elementar, pelo fato dos coeficientes da lei experimental serem valores diferentes dos coeficientes estequiométricos (além disso, reações elementares não podem apresentar coeficientes fracionários na lei).
b) Da lei de velocidade:
k = v/([A]1,50[B]0,50[C]0,25)
Ao substituir os valores de um experimento específico (qualquer um dos quatro fornece o mesmo valor nesse caso em específico), obtém-se que k = 0,0152 (L/mol)1,25 s-1.
c) Da equação de Arrhenius, obtém-se que:
ln(v2/v1) = Ea (T1-1 – T2-1) / R
Ea = R ln(v2/v1)/(T1-1 – T2-1) = R ln(20,4)/[298-1 – 318-1] = 118,8kJ mol-1
d) Da equação de Arrhenius, obtém-se que:
ln(v2/v1) = Ea (T1-1 – T2-1) / R
T2-1 = T1-1 - R ln(v2/v1)/ Ea ⇔ T2 = 312K = 39°C
#36
) A técnica para obter os coeficientes é dividir leis de velocidades de diferentes experimentos, considerando que a constante de velocidade é igual para todos:
Utilizando os experimentos II e III, obtém-se o coeficiente a do reagente A:
(0,230/0,115)a = 0,1370/0,0484 ⇔ a = [log 0,1370/0,0484]/[log 0,230/0,115] = 1,50
Utilizando os experimentos III e IV, obtém-se o coeficiente b do reagente B:
(0,175/0,230)1,50. (0,050/0,030)b = 0,1174/0,1370 ⇔ b = 0,50
Utilizando os experimentos I e IV, obtém-se o coeficiente c do reagente C:
(0,175/0,050)1,50 . (0,050/0,030)c = 0,1174/0,0158 ⇔ c = 0,25
A lei de velocidade experimental é v = k[A]1,50[B]0,50[C]0,25.
Conclui-se que a reação não pode ser elementar, pelo fato dos coeficientes da lei experimental serem valores diferentes dos coeficientes estequiométricos (além disso, reações elementares não podem apresentar coeficientes fracionários na lei).
b) Da lei de velocidade:
k = v/([A]1,50[B]0,50[C]0,25)
Ao substituir os valores de um experimento específico (qualquer um dos quatro fornece o mesmo valor nesse caso em específico), obtém-se que k = 0,0152 (L/mol)1,25 s-1.
c) Da equação de Arrhenius, obtém-se que:
ln(v2/v1) = Ea (T1-1 – T2-1) / R
Ea = R ln(v2/v1)/(T1-1 – T2-1) = R ln(20,4)/[298-1 – 318-1] = 118,8kJ mol-1
d) Da equação de Arrhenius, obtém-se que:
ln(v2/v1) = Ea (T1-1 – T2-1) / R
T2-1 = T1-1 - R ln(v2/v1)/ Ea ⇔ T2 = 312K = 39°C
#37
a) Sistemas abertos são aqueles em que são possíveis trocas tanto de matéria quanto de energia com as vizinhanças. Sistemas fechados permitem troca de energia, mas não de matéria. Já sistemas isolados não permitem qualquer tipo de troca com as vizinhanças.
b) Aberto: fungo vivo (mesmo que de forma seletiva pela intervenção da membrana plasmática e seus canais, há troca de energia e matéria com o exterior)
Fechado: álcool em um termômetro e esporo de bactéria
Isolado: chá em uma garrafa térmica de qualidade impecável
c) – HF se encontra mais ordenado, pois realiza interações intermoleculares muito mais fortes (do tipo ligação de hidrogênio) que tendem a organizar as moléculas
obs: para se ter ideia de quão mais organizado é, o HF predomina na forma de dímeros (HF)2 em temperaturas próximas da ambiente.
– Ne se encontra mais organizado: mesmo devido às fortes interações intermoleculares do tipo ligação de hidrogênio que tendem a organizar as moléculas de amônia, os graus de liberdade para rotação/deformação de ligações tornam as moléculas de NH3 mais desorganizadas
– CaCl2 (s) se encontra mais ordenado, pois partículas no estado sólido apresentam liberdade de movimentação muito menor que na forma de íons solvatados livres
– H2O (l) se encontra mais organizado, pois a liberdade de movimentação das partículas é menor no estado líquido que no estado gasoso.
#38
a) – HBr apresenta maior entropia molar, já que apresenta interações intermoleculares bem mais fracas que aquelas realizadas pelo HF
– NH3 apresenta maior entropia molar, devido ao número muito maior de graus de liberdade de rotação/deformação de ligações, mesmo que apresente interações intermoleculares mais fortes
– CaCl2 (aq) apresenta maior entropia molar, uma vez que íons solvatados têm uma liberdade de movimentação muito maior que partículas no sólido iônico.
– H2O(g) tem uma entropia molar maior, uma vez que gases apresentam maior liberdade de movimentação que líquidos
– O grafite apresenta maior entropia molar: enquanto o diamante apresenta estrutura tridimensional definida e, portanto, mais restringida, o grafite apresenta estruturas laminares bidimensionais que apresentam relativa liberdade de movimento entre si.
b) – A entropia aumenta tremendamente, já que, partindo-se de um mol de composto sólido (de movimentação limitada de partículas), obtêm-se cinco mols de íons, que apresentam uma liberdade de movimento bem maior.
– A entropia aumenta, pois passa-se de um sólido para um gás, que apresenta entropia molar muito maior que aquela do sólido
– A entropia diminui, uma vez que há uma diminuição do número de mols de compostos gasosos (perceba que a água formada está no estado líquido)
c) Um processo reversível pode ser explicado como um processo que pode ser revertido por uma mudança infinitesimal de uma variável (ou, alternativamente, como um processo em que o sistema, a todo momento, encontra-se em equilíbrio).
Um exemplo teórico de processo reversível (uma vez que não há processos puramente reversíveis na realidade) seria a expansão de um gás contra um pistão vertical, empurrado para baixo por uma massa de água: a evaporação extremamente lenta do líquido (pode-se pensar no escape de moléculas a nível atômico para entender esse processo) altera de forma praticamente infinitesimal a massa de água, que permite um aumento infinitesimal do volume de gás, que configuraria um processo reversível.
#39
a) – A entropia aumenta tremendamente, já que, partindo-se de um mol de composto sólido (de movimentação limitada de partículas), obtêm-se cinco mols de íons, que apresentam uma liberdade de movimento bem maior.
– A entropia aumenta, pois passa-se de um sólido para um gás, que apresenta entropia molar muito maior que aquela do sólido
– A entropia diminui, uma vez que há uma diminuição do número de mols de compostos gasosos (perceba que a água formada está no estado líquido)
b) A Primeira Lei determina matematicamente que ΔU = q + w. Tendo isso em mente:
Adiabático: não há troca de calor, logo q = 0. Assim, ΔU = w
Isotérmico: não há variação de energia interna, a qual é dependente da temperatura (que não varia), de modo que ΔU = 0. Assim, q = -w
Isométrico: não há trabalho, uma vez que não há variação de volume, tal que w = 0. Assim, tem-se que ΔU = q
Isobárico: para uma pressão externa constante pext, pode-se dizer que ΔU = q - pext ΔV.
c) Para um processo espontâneo:
ΔSuniverso ≥ 0
ΔSsistema + ΔSvizinhança ≥ 0
Lembrando que ΔS = q/T:
ΔSsis + qviz/T ≥ 0
Porém, qviz = - qsis = -ΔHsis uma vez que a pressão é constante, de modo que:
ΔSsis – ΔHsis /T ≥ 0
Como T>0, pode-se escrever que:
TΔSsis – ΔHsis ≥ 0 (i)
Definindo que G = H – TS, temos que dG = dH – (TdS + SdT). Como a temperatura é constante, SdT = 0. Assim:
dG = dH – TdS
Assumindo as variações de entalpia e entropia do processo independentes da temperatura, pode-se integrar a expressão acima e obter que:
ΔGsis = ΔHsis – TΔSsis (ii)
Comparando (i) e (ii), obtém-se que:
– ΔGsis ≥ 0
ΔGsis ≤ 0 para um processo espontâneo.
d) Um processo reversível pode ser explicado como um processo que pode ser revertido por uma mudança infinitesimal de uma variável (ou, alternativamente, como um processo em que o sistema, a todo momento, encontra-se em equilíbrio).
Um exemplo teórico de processo reversível (uma vez que não há processos puramente reversíveis na realidade) seria a expansão de um gás contra um pistão vertical, empurrado para baixo por uma massa de água: a evaporação extremamente lenta do líquido (pode-se pensar no escape de moléculas a nível atômico para entender esse processo) altera de forma praticamente infinitesimal a massa de água, que permite um aumento infinitesimal do volume de gás, que configuraria um processo reversível.
#40
a) A = NO2 B = Cu(NO3)2 C = Cu(OH)2 D = H2O E = CuO F = CuSO4
Cu + 4 HNO3 → Cu(NO3)2 + 2 NO2 + 2 H2O
Cu(NO)2 + 2 NaOH → Cu(OH)2 + 2 NaNO3
Cu(OH)2 → CuO + H2O
CuO + H2SO4 → CuSO4 + H2O
CuSO4 + Zn → Cu + ZnSO4
b) O gás A (NO2) é um radical: o nitrogênio apresenta um elétron desemparelhado, configuração que é consideravelmente instável. Por essa razão, ocorre a formação de uma ligação entre dois átomos de nitrogênio de moléculas de A, resultando na dimerização, pelo compartilhamento desses elétrons (que se encontram bem mais estáveis agora, por estarem emparelhados).
c) Seja p a pressão de N2O4 no equilíbrio:
Kp = p/(1-2p)2 = 200
Resolvendo a expressão, obtém-se p = 0,476bar. Assim:
P(NO2) = 1 -2.0,476 = 0,048bar e P(N2O4) = 0,476bar ⇔ P(total) = 0,524 bar
X(NO2) = P(NO2) / P(total) = 0,048/0,524 ⇔ X(NO2) = 0,092 e X(N2O4) = 1 – 0,092 = 0,908.
d) Se a cor marrom se intensifica, então o equilíbrio desloca para as moléculas de NO2 separadas (isto é, para reagentes no equilíbrio de dimerização); assim, conclui-se que o sentido inverso é endotérmico e que a reação de dimerização é exotérmica. Sendo exotérmica, é possível afirmar que sua constante de equilíbrio é menor que 200 em uma temperatura maior que 450K (como 650K).
#41
a) A = NO2 B = Cu(NO3)2 C = Cu(OH)2 D = H2O E = CuO F = CuSO4
Cu + 4 HNO3 → Cu(NO3)2 + 2 NO2 + 2 H2O
Cu(NO)2 + 2 NaOH → Cu(OH)2 + 2 NaNO3
Cu(OH)2 → CuO + H2O
CuO + H2SO4 → CuSO4 + H2O
CuSO4 + Zn → Cu + ZnSO4
b) O produto de solubilidade do precipitado C (Cu(OH)2) é dado pela expressão:
Ksp, C = 4.10-12 = [Cu2+][OH-]2
Em água pura, a solubilidade S é:
S.(2S)2 = 4S3 = 4.10-12 ⇔ S = 10-4 mol L-1
Em NaOH 0,5M, a concentração de íons hidróxido é constante, logo a concentração S’ é:
S’.(0,5)2 = 4.10-12 ⇔ S’ = 1,6.10-11 mol L-1
c) Eo = Eo(Cu2+) - Eo(Zn2+) = 0,34 – (-0,76) = 1,10V
ΔG = -RTlnK = -nFEo ⇔ K = exp (nFEo/RT) = exp (2.96485.1,10/8,3145.298,15) ⇔ K = 1,54.1037
O agente redutor é o zinco e o agente oxidante é o sulfato de cobre (F).
d) Da equação de Nernst:
E = Eo – RT.lnQ / nF = Eo – 0,0592.logQ / n
E = 1,10 – 0,0592/2 x log (7,5.10-5/0,05) = 1,10 – 0,0296 log (1,5.10-3)
E = 1,10 – 0,0296 [log(10-3) + log(3) – log(2)] = 1,10 – 0,0296 [-3 + 0,477 – 0,301]
E = 1,10 – 0,0296.(-2,824) = 1,10 + 0,0836
E = 1,184V
#42
a) Pressão de vapor é a pressão exercida pelo vapor em equilíbrio termodinâmico com o líquido que lhe deu origem. É uma medida da tendência de evaporação do líquido.
b) A temperatura de ebulição é definida como a temperatura em que a pressão de vapor do líquido se iguala à pressão do sistema. Por exemplo, a temperatura de ebulição da água a condições normais de pressão é a temperatura em que a pressão de vapor da água equivale àquela da condição normal, isto é, 1 bar.
A panela de pressão possibilita obter pressões superiores à atmosférica local, o que aumenta a temperatura de ebulição da água, permitindo aumentar a temperatura do banho usado (o que leva a um cozimento mais rápido dos alimentos, por exemplo).
c) Sim, faz, uma vez que a adição de sal leva a um abaixamento da temperatura de congelamento da água líquida (efeito crioscópico da adição de soluto), o que possibilita o resfriamento a temperaturas menores (considerando que o gelo se encontre em condições capazes de resfriar o líquido a tais temperaturas).
#43
a) A temperatura de ebulição é definida como a temperatura em que a pressão de vapor do líquido se iguala à pressão do sistema. Por exemplo, a temperatura de ebulição da água a condições normais de pressão é a temperatura em que a pressão de vapor da água equivale àquela da condição normal, isto é, 1 bar.
A panela de pressão possibilita obter pressões superiores à atmosférica local, o que aumenta a temperatura de ebulição da água, permitindo aumentar a temperatura do banho usado (o que leva a um cozimento mais rápido dos alimentos, por exemplo).
b) Sendo π a pressão osmótica e M a massa molar média do polímero:
πV = nRT = mRT/M ⇔ M = mRT/πV
M = 1,7452.0,08205.298,15/0,162.0,250 ⇒ M = 1054 g mol-1
Sim, esse valor é adequado, pois se encontra na faixa descrita.
c) Seja σ o erro do valor teórico de 1000 g mol-1. Tem-se que:
σ = (Mexp - Mteo) / Mteo = (1054-1000) / 1000
σ = 54/1000 = 0,054 ⇒ σ = 5,4%.
d) Sabe-se que:
ΔT = Kiω = – (Tf’ – Tf) ⇔ Tf = Tf’ + Kiω
Tf’ = 1,403°F = -16,998°C
ω = n/msol = (mpol/M) / (dX.VX) = (1,3420/1054,1) / (0,740.0,4) = 4,301.10-3 mol kg-1
Tf = Tf’ + Kiω = -16,998 + 0,37.1.4,301.10-3 ⇒ Tf = -16,996°C.
Note que como a solução é muito diluída, não há real variação da temperatura de congelamento. Isso poderia ter sido explorado na resposta (o valor de 1,403°F poderia ser usado corretamente como resposta, se seguido por essa justificativa).
#44
a) Sendo π a pressão osmótica e M a massa molar média do polímero:
πV = nRT = mRT/M ⇔ M = mRT/πV
M = 1,7452.0,08205.298,15/0,162.0,250 ⇒ M = 1054 g mol-1
Sim, esse valor é adequado, pois se encontra na faixa descrita.
b) Seja σ o erro do valor teórico de 1000 g mol-1. Tem-se que:
σ = (Mexp - Mteo) / Mteo = (1054-1000) / 1000
σ = 54/1000 = 0,054 ⇒ σ = 5,4%.
c) Sabe-se que:
ΔT = Kiω = – (Tf’ – Tf) ⇔ Tf = Tf’ + Kiω
Tf’ = 1,403°F = -16,998°C
ω = n/msol = (mpol/M) / (dX.VX) = (13,4420/1054,1) / (0,740.0,2) = 8,616.10-2 mol kg-1
Tf = Tf’ + Kiω = -16,998 + 2,720.1.8,616.10-2 ⇒ Tf = - 16,764°C = 256,39K
d) Da lei de Raoult:
Pv = Pvo .X
Em que X é a fração molar de solvente na solução, tal que X = nsolvente / (nsolvente + nsoluto)
Note, contudo, que a quantidade de soluto é substancialmente menor que a de solvente, de modo que se pode aproximar que X≈1.
Assim, Pv, 363,15o = 0,5933 bar.
Sabe-se também que, a 405,8K, Pvo405,8 = 1,0000 bar. Usando-se a equação de Van’t Hoff:
ln (Pvo405,8 / Pvo363,15) = ΔHvap . (363,15-1 – 405,8-1) / R
ΔHvap = R. ln (Pvo405,8 / Pvo363,15) / (363,15-1 – 405,8-1)
ΔHvap = 8,3145. ln (1,0000/0,5933) / (363,15-1 – 405,8-1)
ΔHvap = 15,00 kJ mol-1
OBS: Além de ser eficiente e poupar tempo de resolução, aproximações válidas como esta são por vezes obrigatórias para a resolução – perceba que não há dados sobre a massa molar de X, de modo que é impossível resolver propriamente essa questão sem alguma aproximação. Há, pois, uma única possibilidade: aproximar que X≈1 (estimar um valor para a massa molar do solvente X carrega consigo um erro extremamente maior e não é uma forma válida de resolução).
e) Pela regra de Trouton:
Teb≈ ΔH/ ΔSvapmédio = 15,00.103/85 = 176,47 K
σ = (Tebest – Tebreal) / Tebreal = (176,47 – 405,8)/405,8 = - 56,5%
O cálculo indica que o líquido X apresenta um valor de temperatura de ebulição muito superior ao estimado pela regra de Trouton. Isso mostra que a variação de entropia do processo de vaporização é bem menor que aquela esperada para um líquido médio como o benzeno – o que pode indicar uma desordem muito maior que a média no estado líquido (fracas interações intermoleculares) ou uma ordem muito maior no estado gasoso (como a formação de dímeros, trímeros ou afins).
Chega-se a essa conclusão pela análise da Regra de Trouton: para uma substância com baixo valor de ΔHvap como a substância X, espera-se um baixo ponto de ebulição; como esse ponto é muito maior que o esperado, conclui-se que o processo é menos termodinamicamente favorecido entropicamente do que o esperado, por conseguinte.
#45
a) Não, não está, uma vez que no cátodo ocorre redução (recebimento de elétrons), que reduz o NOx da espécie envolvida. O NOx aumenta no ânodo, onde ocorre a oxidação.
b) Não, não é sempre válida, uma vez que na eletrólise os polos são invertidos (isto é, o cátodo é negativo e o ânodo é positivo); assim, a afirmação de que “o anodo é negativo” é válida apenas para pilhas.
c) Note-se que a solução de A reagiu com todas as barras, o que mostra uma forte tendência a redução do cátion do metal A, que por sua vez indica um alto valor de potencial de redução. Assim, conclui-se que A é o paládio, Pd.
Já a solução de B não reage com nenhuma barra, o que indica forte tendência de manter-se na forma oxidada catiônica. Isso indica um valor muito negativo de potencial de redução, de modo que B é o magnésio, Mg.
A solução de D reage com todas as barras, exceto a de paládio, o que indica que possui o segundo maior potencial de redução. Assim, conclui-se que D é o chumbo, Pb.
A solução de E reage apenas com a barra de magnésio, de modo que apresenta o segundo menor potencial de redução. Assim, o metal E é o zinco, Zn.
Por exclusão, o metal C é o níquel, Ni.
Todas as reações são do tipo M2+ + Me → M + Me2+.
#46
a) Substituindo os valores na expressão dada, obtemos:
logkp= -4374/573 + 1,75 log573 + 3,78
kp= 100,973 = 9,40
Usando a relação Kp = Kc (RT)Δn :
9,397 = Kc (0.082 . 573)2-1 >>> Kc = 0,20
b) Sabemos que a soma das pressões equivale a 150 kPa, ou seja, 1,5 bar.
P(PCl5) + P(PCl3) + P(Cl2) = 1,5 >>> Sabemos que P(PCl3)= P(Cl2), pois ambos não existiam no recipiente e foram formados em mesma proporção. Agora, chamemos P(PCl3)= P(Cl2) = X
Substituindo na expressão do Kp obtemos:
Kp= 0,2 = X2/(1,5 - 2X) >>> X2 + 0,400X - 0.3 = 0 >>> X = 0,383
Portanto, P(PCl3)= P(Cl2) = 0,383 bar e P(PCl5) = 0,734 bar.
c) Tomemos como referência um valor qualquer de quantidade de matéria da espécie PCl5 como, por exemplo, 1 mol.
Sabemos que n(PCl3)= 1- n(PCl5) e que, em uma situação ideal, a razão entre o número de mols de gás é igual a razão entre as pressões. Daí, obtemos a expressão:
(1-nPCl5)/ nPCl5 = 0,383/ 0,734 >>> nPCl5 = 0,657 e nPCl3= 0,343
Podemos definir o grau de dissociação, α, como sendo a razão entre a parte dissociada e a parte inicial, portanto:
α = nPCl3/n0PCl5 >>> α= 0,343/1 >>> α= 34,3 %
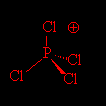
d) Os íons que compõe o sólido iônico são PCl4+ e PCl6-. Suas respectivas estruturas de lewis são:
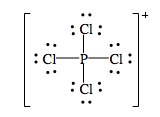

E suas geometrias são:
 Tetraédrica
Tetraédrica
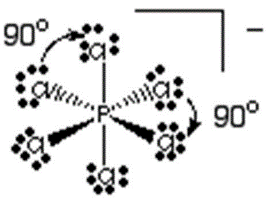 Bipirâmide de base quadrada
Bipirâmide de base quadrada
e) O espectro obtido pelo método de ressonância magnética nuclear mostra diferentes sinais para átomos em diferentes ambientes químicos. Ou seja, como apenas um sinal é obtido, todos os átomos de flúor ocupam o mesmo ambiente químico. A explicação para isso é que ocorre uma rápida alternância entre os átomos de flúor das posições axiais e equatoriais, o que faz com que todos eles sejam equivalentes.

#47
Primeiro calcularemos o K a 1200°C:
![K = RT * \dfrac{[F]^2}{[F_2]} \Rightarrow K = 33](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_52fe166ca81b09fc3603b7bfa4225eb7.gif?ssl=1)
Depois converteremos ele para o K a 25°C:


Por ultimo finalizaremos usando  :
:

#48
a) Sabendo a massa inicial, a massa final e a meia-vida, é possível calcular o tempo que passou entre a medição da massa final e da massa inicial. Para seres vivos isso é extremamente eficiente pois o ser humano, por exemplo, está trocando matéria com o meio ambiente constantemente como o carbono-14, logo a quantidade de carbono-14 no corpo humano é aproximadamente constante e a porcentagem de carbono-14 que existe no mundo. Logo a massa inicial é conhecida. Como quando o ser humano morre ele para de trocar matéria com o meio ambiente, a quantidade de carbono-14 vai diminuir na sua taxa normal que é igual a meia vida. Logo medindo a massa final, podemos descobrir a quanto tempo um ser vivo morreu.
b) Como  , o tempo decorrido foi 28 meias-vidas, se o tempo decorrido foi 7000 anos,
, o tempo decorrido foi 28 meias-vidas, se o tempo decorrido foi 7000 anos, 
c) 
d)
A concentração é: ![[X]_0 = \dfrac{\frac{m}{M}}{V} \Rightarrow [X]_0 = \dfrac{\frac{500*10^{-3}}{500}}{1} \Rightarrow [X]_0 = 10^{-3}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_ebdfe0596c5f822c8b0b2dd34ebf6625.gif?ssl=1)
Para segunda ordem: ![t_{1/2} = \dfrac{1}{k*[X]_0} \Rightarrow k = 4 L \, ano^{-1} \, mol ^{-1}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5c606aaff1eaa621a3787a2fb760c51b.gif?ssl=1)
#49
A)

B)

C)

D) i) O sinal negativo indica que a entropia do sistema diminuiu, ou seja, encontra-se mais organizado.
ii) Essa maior organização é explicada pela presença de carga, pois, ao ionizar-se, a água forma espécies não neutras. Tais espécies promovem interações intermoleculares mais intensas, o que aproxima as moléculas, deixando-as mais organizadas e, por conta disso, a entropia do sistema diminui.
#50
1)

2)

3)I) Sabe-se que a atividade de um sólido puro é nula, portanto, uma variação em sua quantidade não atingiria o quociente reacional e, dessa forma, não alteraria a voltagem da pilha.
II)

Note que ao usarmos a equação der nernst fica evidente a diminuição do potencial dessa semi-reação com o acréscimo da concentração de íons zinco. Portanto, ao dobrar a concentração de íons zinco há diminuição do potencial da pilha.
#51
- Uma bomba nuclear funciona tendo como princípio a fissão nuclear. Em uma reação de fissão nuclear, um núcleo atômico se divide em dois núcleos mais leves, liberando bastante energia no processo. Com o urânio uma reação de fissão é a seguinte:
23892U+10n->9836Kr+14156Ba+310n
O interessante de se notar nessa reação é que esses três nêutrons podem ser usados para iniciar outra reação de fissão como essa, gerando um número exponencial de fissões e como essa reação libera bastante energia, isso gera uma explosão
- Tanto o boro quanto o carbono são materiais que absorvem nêutrons, e como a reação de fissão requer nêutrons, caso materiais como esses forem colocados no meio eles desaceleram a reação de fissão
- Vamos calcular o ΔH da reação a partir da fórmula de Einstein:
238U->α+234Th
ΔE=Δm.c2
ΔH=(234,0436+4,0026-238,0508).(2,998*108)2/103=-4,134*108 kJ/mol
- Combustão do octano:
C8H18+12,5O2->8CO2+9H2O
ΔH=∑ΔfHprodutos-∑ΔfHreagentes=-5494,5kJ/mol
Aniquilação do elétron:
e-+e+->energia
ΔE=Δm.c2
ΔH=(2*9,1*10-31).(2,998*108)2*6,02*1023=-9,85*108 kJ/mol
#52
Obs: com uma pesquisa na internet podemos encontrar que:
Ce4++Fe2+->Ce3++Fe3+ Eo=+0,67V
Vamos calcular a constante de equilíbrio da reação. ΔG=-nFE=-RTlnK, logo K=2,14*1011. Por isso durante a questão, quando devido, vamos considerar que a reação foi completa,
a) A quantidade de Fe2+ adicionada é menor que a quantidade de Ce4+, por isso, podemos assumir que todo o Fe2+ foi consumido. A quantidade de Ce3+ formado é :
nCe3+=nFe2+=C*V=0,01*10*10-3=10-4mol
e a quantidade de Ce4+ é:
nCe4+=n0-nCe3+=4*10-4
Usando a equação de Nernst:
E=Eo-RTlnQ/(nF), sendo Q=[Ce3+]/[Ce4+]
encontramos que E=1,48V
b) Esse é o ponto de equivalência. Nesse ponto não é válido assumir que a reação se processou até a completude pois geraria [Ce4+]=[Fe2]=0 que não satisfaria a constante de equilíbrio. Da estequiometria da reação sabemos que:
[Ce3+]/[Ce4+]=[Fe3+]/[Fe2+].
Podemos calcular o potencial da reação por cada uma das semirreações. Reação do Ferro:
E=EoFe2+/Fe3+-RTln([Fe2+]/[Fe3+])/nF
Reação do Cério:
E=EoCe4+/Ce3+-RTln([Ce3+]/[Ce4+])/nF
Somando as equações:
2E= EoCe4+/Ce3+- EoFe2+/Fe3+-RTln([Ce3+][Fe2+]/[Ce4+]*[Fe3+])/nF
Usando da estequiometria do ponto de equivalência:
E=( EoCe4+/Ce3++EoFe2+/Fe3+)/2=1,11V
c) Neste ponto há um excesso de Fe2+, por isso vamos usá-lo para calcular o potencial. A quantidade de Fe3+ é:
nFe3+=n0,Ce4+=5*10-4
A quantidade de Fe2+ é:
nFe2+=C*V-nFe3+=2*10-4
Equação de Nernst:
E=Eo-RTlnQ/(nF), sendo Q=[Fe2+]/[Fe3+]
Encontramos E=0,79V
d) O potencial da reação varia de aproximadamente 1,5 até 0,75V. Pelo diagrama as espécies possíveis são: MnO4-, MnO2 e Mn2+
#53
Item d
#54
O ideal para encontrarmos os valores de  no processo é calcularmos esses valores para cada etapa e depois somá-los. Para isto devemos encontrar a temperatura no início da etapa
no processo é calcularmos esses valores para cada etapa e depois somá-los. Para isto devemos encontrar a temperatura no início da etapa  , onde podemos achá-la por meio da lei de Charles:
, onde podemos achá-la por meio da lei de Charles:

: Na etapa nós temos que o processo ocorre em volume constante, decorrente deste fato podemos concluir que o  é igual a zero e também que o
é igual a zero e também que o  é igual ao
é igual ao  . Com isto em mente é possível calcular o ; e o
. Com isto em mente é possível calcular o ; e o  da seguinte forma:
da seguinte forma:


Resumo dos valores da etapa :

 : Na etapa nós temos que o processo ocorre sem troca de calor com a vizinhança, decorrente deste fato podemos concluir que o
: Na etapa nós temos que o processo ocorre sem troca de calor com a vizinhança, decorrente deste fato podemos concluir que o  é igual a zero e também que o
é igual a zero e também que o  é igual ao
é igual ao  . Com isto em mente é possível calcular o ;
. Com isto em mente é possível calcular o ;  e o
e o  da seguinte forma:
da seguinte forma:


Resumo dos valores da etapa :

Processo total: Neste processo nós podemos somar os valores das etapas e para acharmos os valores do processo total. É possível fazer isso normalmente com o  e o
e o  pois eles são propriedades de estado, ou seja, independente do caminho que eles façam, se tiver mesmo estado inicial e final os valores deles sempre serão iguais. Já com o e o
pois eles são propriedades de estado, ou seja, independente do caminho que eles façam, se tiver mesmo estado inicial e final os valores deles sempre serão iguais. Já com o e o  , nós só podemos fazer isto decorrente que em etapas alternadas do processo descrito o valor deles foi igual a zero, de maneira que restasse apenas a possibilidade de calculá-los em determinadas etapas ao invés de calcularmos no processo como um todo. Desta forma concluímos que os resultados para o processo total serão iguais a:
, nós só podemos fazer isto decorrente que em etapas alternadas do processo descrito o valor deles foi igual a zero, de maneira que restasse apenas a possibilidade de calculá-los em determinadas etapas ao invés de calcularmos no processo como um todo. Desta forma concluímos que os resultados para o processo total serão iguais a:
 ;
;  ;
;  ;
; 
#55
Para essa questão devemos lembrar que cada grau de movimento de uma partícula tem uma contribuição de  para a energia interna, desta forma devemos lembrar das estrutura de cada composto nos sistemas descritos para assim determinamos os graus de movimento existentes em cada um, para depois somá-los. Sabendo que
para a energia interna, desta forma devemos lembrar das estrutura de cada composto nos sistemas descritos para assim determinamos os graus de movimento existentes em cada um, para depois somá-los. Sabendo que  , temos que os termos serão substituídos por
, temos que os termos serão substituídos por  , pois em todos os sistemas temos a presença de
, pois em todos os sistemas temos a presença de  de moléculas e inicialmente iremos calcular o valor para cada partícula. Sabendo que
de moléculas e inicialmente iremos calcular o valor para cada partícula. Sabendo que  é o grau de movimento translacional e que
é o grau de movimento translacional e que  é o grau de movimento rotacional, temos que a contribuição por mol de cada um será:
é o grau de movimento rotacional, temos que a contribuição por mol de cada um será:
 por molécula
por molécula por mol
por mol
 por molécula
por molécula por mol
por mol
 por molécula por mol
por molécula por mol
 por molécula
por molécula por mol
por mol
 por molécula
por molécula por mol
por mol
Substituindo os valores e alocando os itens em comum em evidência, temos:





Desta forma concluímos que a sequência crescente correta dos itens seria:

#56
a) Como a transformação de A para B é isobárica,  . Assim, utilizando a Lei dos Gases ideais:
. Assim, utilizando a Lei dos Gases ideais:

Portanto, as coordenadas de A são:

Para as coordenadas de B, sabe-se que:

Assim, fazendo uma substituição, utilizando a Lei dos Gases ideais, podemos reescrever como:

Logo:

Aplicando esse valor na equação original:


Portanto, as coordenadas de C são:

b) Não é possível saber nenhuma informação direta do calor trocado na transformação de A para B, mas é possível descobri-lo utilizando a Primeira Lei da Termodinâmica.
Como U é uma função de estado, sua variação não depende do caminho feito. Assim, a variação de Energia interna de A para A, deve ser a mesma que de A para B, de B para C e de C para A novamente. Ou seja:

 , pois é uma transformação isoterma. Para as outras transformações podemos reescrever a
, pois é uma transformação isoterma. Para as outras transformações podemos reescrever a  por
por  em cada caso e isolar o calor de AB. Ou seja:
em cada caso e isolar o calor de AB. Ou seja:

 , pois a transformação é adiabática.
, pois a transformação é adiabática.  é fácil de calcular, pois é um trabalho a pressão constante, ou seja:
é fácil de calcular, pois é um trabalho a pressão constante, ou seja:  . Já sabemos o valor de
. Já sabemos o valor de  e
e  pela questão anterior e assim podemos calcular:
pela questão anterior e assim podemos calcular:

O mais difícil de se calcular é  , pois aqui é necessário o uso de Cálculo. Sabe-se que a forma geral de um trabalho ao longo de uma pressão não constante é:
, pois aqui é necessário o uso de Cálculo. Sabe-se que a forma geral de um trabalho ao longo de uma pressão não constante é:  .
.
Para saber o trabalho de BC precisamos então escrever o P nessa transformação em função de uma variável V e fazer uma integral definida dessa função indo de B para C.
A única informação que temos sobre a relação de P e V ao longo dessa adiabática é de que o valor  é constante. Chamando essa constante de um certo
é constante. Chamando essa constante de um certo  , podemos reescrever a pressão como:
, podemos reescrever a pressão como:  . Integrando isso de B para C:
. Integrando isso de B para C:

Porém, lembre-se que havíamos definido Z como a constante de  ao longo da adiabática e ambos esses pontos pertencem à essa adiabática. Ou seja,
ao longo da adiabática e ambos esses pontos pertencem à essa adiabática. Ou seja,  . Substituindo no valor do trabalho calculado, temos que:
. Substituindo no valor do trabalho calculado, temos que:

Colocando os valores na outra fórmula, temos que:

c) A Entalpia é definida como:

Entalpia também é uma função de estado, logo sua variação depende apenas do estado inicial e do estado final. Assim, podemos escrever que, para qualquer transformação:

Como no item a) calculamos P e V para todos os pontos e no item b) calculamos para todas as transformações, poderíamos simplesmente aplicar esses valores para cada transformação e ter a resposta. Esse jeito seria possível, porém um pouco mais trabalhoso. Utilizando algumas propriedades da entalpia podemos resolver isso de um jeito mais rápido, mas as respostas serão as mesmas que calculando formalmente.
Na transformação CA,  e PV é constante ao longo da transformação, logo:
e PV é constante ao longo da transformação, logo:

Na transformação AB, a pressão é constante. Assim, o termo  . Porém, esse termo é exatamente o oposto do trabalho realizado nessa transformação. Dessa forma o termo de trabalho e o termo de PV se cancelam e a entalpia é igual ao calor dessa transformação. Isso é uma propriedade importante da entalpia: a pressão constante, ela é sempre igual ao calor. Nós calculamos o calor de AB no item B, logo:
. Porém, esse termo é exatamente o oposto do trabalho realizado nessa transformação. Dessa forma o termo de trabalho e o termo de PV se cancelam e a entalpia é igual ao calor dessa transformação. Isso é uma propriedade importante da entalpia: a pressão constante, ela é sempre igual ao calor. Nós calculamos o calor de AB no item B, logo:

Como entalpia é uma função de estado, podemos fazer o mesmo que fizemos para a energia interna no item b) para isolar a entalpia de BC:


Substituindo os valores de entalpia já calculados:

#57
a)  Reação de
Reação de  ordem
ordem
b) 
c) 
#58
a)


b)
Conforme a pressão aumenta, o grau de dissociação diminui.
Conforme a temperatura aumenta, o grau de dissociação aumenta.
#59
a)


b)



c)


d)




#60
Esse problema trata sobre o deslocamento do equilíbrio seguindo o Principio de Le Chatelier, então devemos sempre procurar a saída do sistema para tentar reverter o efeito da ação externa.
a) Como a reação direta é exotérmica, o aumento da temperatura deslocaria o equilíbrio para o lado dos reagentes, visto que sua produção é endotérmica, o que se favorece com o aumento da temperatura.
b) Para diminuir a pressão nesse caso, será necessário aumentar o volume do sistema, por conta disso o equilíbrio se deslocará para o lado que tiver maior volume gasoso (maior número de mols), dessa maneira na seguinte reação seria deslocado para o lado dos reagentes.
c) Como foi adicionado mais reagentes, o valor do quociente reacional se torna menor do que o da constante de equilíbrio, dessa maneira o sistema tenderá a produzir mais produtos afim de alcançar o uma nova posição de equilíbrio.
d) Como o gás não irá reagir com nenhum dos componentes, sua concentração não irá ser afetada, logo o equilíbrio não será afetado.
e) Nesse caso, como o volume foi alterado deve se considerar o caso em que ele aumenta e o caso em que ele diminui, se o volume aumentar o equilíbrio se deslocará para o lado de maior volume gasoso, nesse caso o dos reagentes, já se for diminuído se deslocará para o lado dos produtos.
#61
 Comparando o
Comparando o  com
com  , notamos que há uma diferença de 4 no número de massa e 2 no número atômico. Logo, a reação é da forma:
, notamos que há uma diferença de 4 no número de massa e 2 no número atômico. Logo, a reação é da forma:

 +
+ 
Sabemos que a partícula corresponde ao núcleo de hélio que é um gás nobre o que produz uma pressão maior no interior do recipiente. Assim, sabendo que a reação é de 1:1 com relação aos átomos consumidos de bismuto e produzidos de partículas , respectivamente:
\begin{equation*}
PV=nRT, n=\frac{PV}{RT}
\end{equation*}
\begin{equation*}
\frac{386atm.10L}{0,082LatmK^{-1}mol^{-1}.298K}=n=158,5mol \end{equation*}
A massa molar do bismuto é  , logo:
, logo:
\begin{equation*}
158,7mol.214g/mol=33,9.10^3g\end{equation*}
 Como a reação é de primeira ordem, valem as relações:
Como a reação é de primeira ordem, valem as relações:
\begin{equation*}
ln\frac{m}{m_0}=-kt , t_{1/2}=\frac{ln2}{k}\end{equation*}
Rearranjando obtemos:
\begin{equation*}
-\frac{t_{1/2}.ln\frac{m}{m_0}}{ln2}=t\end{equation*}
Substituindo e tendo em vista que a massa de Bismuto que irá ter no momento em que a quantidade de helio é suficiente para estourar o vidro será  :
:
\begin{equation*}
-\frac{19,9min.ln\frac{26,1kg}{60kg}}{ln2}=t=23,9 min\end{equation*}
#62
a) Primeiro, deve-se ter em mente que para um líquido entrar em vaporização, ele necessita ter a pressão de vapor igual ou superior a pressão atmosférica já que ele necessita vencer a força que atua sobre sua superfície. Assim, basta analisarmos o ponto do gráfico onde a altura corresponde à altura dos dois locais em relação ao nível do mar. Para Curitiba, o ponto é aquele em que a ordenada é aproximadamente  . Para a Cordilheira dos Andes, o ponto é aquele em que a ordenada é aproximadamente
. Para a Cordilheira dos Andes, o ponto é aquele em que a ordenada é aproximadamente  . Veja a figura abaixo:
. Veja a figura abaixo:

b) Com base no item a, já sabemos que o a água precisa ter uma pressão de vapor maior para entrar em ebulição em Curitiba e, portanto, precisa de mais energia para fazer isso. Assim, o local que ela menos precisaria de calor para entrar em ebulição é na Cordilheira dos Andes.
c) Para este processo, a água líquida não deve participar da expressão da constante  , de modo que o processo de vaporização bem descrito da forma:
, de modo que o processo de vaporização bem descrito da forma:
\begin{equation}
K_p=P_{H2O(g)}\end{equation}
d) Pela equação de Van 't Hoff, temos que:
\begin{equation}
ln(K)=\frac{\Delta S}{R} - \frac{\Delta H}{RT}\end{equation}
Substituindo em (1) em (2):
\begin{equation}
ln(P_{H2O(g)})=\frac{ \Delta S }{R} - \frac{\Delta H}{RT}\end{equation}.
Assim deduzimos a equação de Clausius-Clapeyron:
\begin{equation*}
ln(P_1)-ln(P_2)=\frac{\Delta S}{R}-\frac{\Delta S}{R}-\frac{\Delta H}{RT_1}-\left(-\frac{\Delta H}{RT_2}\right)\end{equation*}
\begin{equation}
ln\left(\frac{P_1}{P_2}\right)=-\frac{\Delta H}{R}\left( \frac{1}{T_1}-\frac{1}{T_2}\right).\end{equation}
e) Como a densindade da água é  , sabemos que a massa de água presente no sistema é
, sabemos que a massa de água presente no sistema é  ou
ou  . Assim, o número de mols de água presente será a massa de água dividido pela massa molar da água:
. Assim, o número de mols de água presente será a massa de água dividido pela massa molar da água:
\begin{equation*}
n= \frac{m_{\acute{a}gua}}{MM_{\acute{a}gua}}=\frac{1000 g}{18 g/mol} \approx 55,6 mol\\\end{equation*}
Agora calculando o valor da energia total fornecida para a ebulição da água:
\begin{equation*}
P=\frac{Q}{\Delta t} \leftrightarrow P.\Delta t=Q\end{equation*}
Onde P é a pontência em watts fornecida pelo sistema,  o tempo em segundos e Q o calor em joules. Como
o tempo em segundos e Q o calor em joules. Como  =
= , temos que:
, temos que:
\begin{equation*}
\Delta H=\frac{\Delta t.P}{n}=\frac{5.60 s . 8,1 kW}{55,6 mol}=43,7 kJ/mol.\end{equation*}
f) Com base na equação de Clausius-Clapeyron (4), podemos calcular o valor da temperatura de ebulição:
Para Curitiba, sabemos que a pressão de vapor é  (figura 1). Portanto, substituindo:
(figura 1). Portanto, substituindo:
\begin{equation*}
ln\left(\frac{1 atm}{0,9 atm}\right)=-\frac{43700 J}{8,314 J/K.mol}\left(\frac{1}{373 K}-\frac{1}{T_1}\right) , T_1= 370,23 K= 97.23^\circ C.\end{equation*}
Para os Andes, sabemos que a pressão de vapor necessária é  . Substituindo:
. Substituindo:
\begin{equation*}
ln\left(\frac{1 atm}{0,4 atm}\right)=-\frac{43700 J}{8,3144621 J/K.mol}\left(\frac{1}{373 K}-\frac{1}{T_2}\right) , T_2= 350,36 K= 77,21^\circ C.\end{equation*}
#63

 é a Energia Livre de Gibbs,
é a Energia Livre de Gibbs,  é a quantidade de eletrons,
é a quantidade de eletrons,  é a Constante de Faraday e
é a Constante de Faraday e  é o potencial de redução padrão.
é o potencial de redução padrão. (
( )
)
 (
( )
)
___________________________________________________________________________________________
 (
( )
)




B)



C)
O ânodo é onde a oxidação acontece, ou seja: 
O cátodo é onde a redução acontece, ou seja: 
O diagrama da pilha é: 
D)
Estudando a eletrólise do ânodo, temos:

Apartir desta semi-reação, tiramos a relação:
 de elétrons x
de elétrons x 

 de vanádio
de vanádio
 Massa de vanádio
Massa de vanádio
Massa de vanádio = 
Massa de vanádio = 
#64
Para a resolução dessa questão devemos pensar nos processos possíveis de acontecer nesse cenário:
1° processo - Aquecimento do gelo até 273K -> 
2° Processo - Fusão do gelo -> 
3° Processo - Resfriamento da água a 293K até a temperatura de equilíbrio -> 
4° Processo - Aquecimento da água a 273K até a temperatura de equilíbrio -> 
a) Após listar os processos é apenas necessário aplicar que a soma dos calores deverá sempre ser 0.
Dica: Perceba que os valores das temperaturas em Celsius serão mais simples de serem trabalhados e como estamos trabalhando com variação, não há problema em trocarmos a unidade.
Dessa maneira, temos que:




b) As variações de entropia devem ser calculadas por etapa e depois somadas, cada uma correspondendo a um processo em específico.





#65
 , pois o
, pois o  apresenta pontes de hidrogênio que fazem com que sua estrutura seja mais organizada do que que apresenta interações intermoleculares do tipo dipolo-dipolo. Lembre-se que interações do tipo dipolo-dipolo são mais fracas que as pontes de hidrogênio.
apresenta pontes de hidrogênio que fazem com que sua estrutura seja mais organizada do que que apresenta interações intermoleculares do tipo dipolo-dipolo. Lembre-se que interações do tipo dipolo-dipolo são mais fracas que as pontes de hidrogênio.
 , pois a energia cinética média das moléculas no estado sólido é sempre menor que no estado líquido para uma mesma pressão. Assim a desordem no estado líquido é maior.
, pois a energia cinética média das moléculas no estado sólido é sempre menor que no estado líquido para uma mesma pressão. Assim a desordem no estado líquido é maior.

 a
a  , pois quanto mais espaço as moléculas tiverem para estar, maior a entropia do sistema e, pela equação de Clapeyron, quanto maior o espaço, menor a pressão do sistema. Assim, concluímos que a entropia depende do inverso da pressão e, consequentemente, a tem maior entropia que a
, pois quanto mais espaço as moléculas tiverem para estar, maior a entropia do sistema e, pela equação de Clapeyron, quanto maior o espaço, menor a pressão do sistema. Assim, concluímos que a entropia depende do inverso da pressão e, consequentemente, a tem maior entropia que a  .
.
#66
Escrevendo as reações da pilha sabendo que, como o cobre tem potencial de redução maior, este reduz:
Reação catódica: 
Reação anódica: 
Reação global: 
Assim, podemos tirar que, pela equação de Nernest:
![E=E^0-\frac{0,0592}{n}.log(\frac{[Zn^{2+}]}{[Cu^{2+}]})](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_ad6ad92918656b758622018082eb84af.gif?ssl=1)
Portanto, fazendo as contas: 
Agora, sabendo que U=R.I onde U é a ddp em volts, R é a resistência em ohms e I é a corrente elétrica em ampéres:

Portanto, I=0,094 A=0,094 C/s.
Pela constante de Faraday:
 de elétrons.
de elétrons.
Para cada mol de eletros temos 0,5 mols de Cobre pela estequiometria. Portanto, temos  mol/s de cobre. Multiplicando pela massa molar do cobre:
mol/s de cobre. Multiplicando pela massa molar do cobre:  de cobre.
de cobre.
#67
a) 
b) Todos os outros restantes: 






c) Tais identificações podem ser feitas lembrando-se do princípio de Le Chatelier que diz que o aumento da temperatura sempre favorecerá o lado endotérmico da reação, dessa maneira fica possível concluir que aqueles sais cuja a solubilidade aumenta com a temperatura possuem dissolução endotérmica e os outros exotérmicas.
#68
a) Pela lei de Henry,  . Então:
. Então: ![[CO_2]=2,3.10^{-4}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_699c2d3ca958afab87ddd9ac551a8598.gif?ssl=1)
 . Assim:
. Assim:
![[CO_2]=1,22.10^{-3}M](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_6099ce0f82410bfd5e6689b6fb018794.gif?ssl=1)
b) Como se trata de um tampão entre o dióxido de carbono e o bicarbonato, utilizaremos a equação de Handersson-Hasselbach:

Substituindo os valores:
![7,4=6,1+log(\frac{[HCO_3^-]}{[CO_2]})](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_42a5b7d7b1ae74d68320e8b7f7775a62.gif?ssl=1) .
.
Daí:
![[HCO_3^-]=0,024mol/L](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_ddf6dd4155ec03994af9940eecf3560f.gif?ssl=1) .
.
#69
O catalisador é utilizado para ser criado um novo caminho reacional com menor energia de ativação, porém não altera as funções de estado ou a posição de equilíbrio.
a) Não, pois é função de estado.
b) Não, pois é função de estado.
c) Não, pois é função de estado.
d) Não, pois a posição de equilíbrio é constante para uma reação a uma temperatura constante.
#70
a) Um aumento na pressão faz com que o equilíbrio tenda a um menor volume devido à energia mínima do sistema deslocando, assim, o equilíbrio para o lado que tem o menor número de mol de gases da reação. Portanto, desloca o equilíbrio para o lado dos produtos.
b) Como a reação é exotérmica, um aumento da temperatura tenderá à formação de reagentes pelo princípio de Le Châtelier.
c) A adição de gás inerte não causa deslocamento do equilíbrio.
#71
A adição de agentes complexantes com o cátion do sal, faz com que uma energia de estabilização seja gerado na interação metal-ligante, fazendo com que a solubilidade do sal seja aumentada.
#72

Visivelmente, o  para essa reação é de -2, assim temos que:
para essa reação é de -2, assim temos que:


#73
a)

O valor 0 da entalpia é atribuído, por convenção, às substâncias simples em sua forma mais estável, como o carbono e o silício estão em suas formas alotrópicas mais estavéis, eles possuem  . Assim:
. Assim:



b)
A variação de entropia é dada por:

Então:

Substituindo os valores dados:


c)
A energia livre de Gibbs é dada por:

Como já calculamos a entropia da reação, apenas precisamos substituir na equação da energia livre de Gibbs.


d)
Para uma reação ser espontânea, o seu  precisa ser menor do que zero. Logo:
precisa ser menor do que zero. Logo:

Isolando T:



Assim, verificamos que T precisa ser maior que  para que a reação de produção do silicone seja espontânea
para que a reação de produção do silicone seja espontânea
#74
Para solucionarmos essa questão, é necessário descobrirmos o fator de crescimento da constante original do método 2. Esse processo será dividido em duas etapas (Considere: Sem índice para reação original, índice para reação do método e índice para reação do método ).
Etapa I: Busca pela energia de ativação da reação original, fazendo-o por meio da equação de Arrhenius para a constante de reação:





Etapa II: Utilizar a energia de ativação na fórmula de arrhenius para a mudança da constante conforme a variação de temperatura, com intuito de acharmos o fator de crescimento da constante original do método : (O  encontrado na etapa anterior será representado por
encontrado na etapa anterior será representado por  na conta, sendo ele guardado na memória da calculadora para fornecer o resultado mais exato possível).
na conta, sendo ele guardado na memória da calculadora para fornecer o resultado mais exato possível).



Com isso, podemos concluir que haverá um maior aumento na velocidade de reação se o engenheiro químico preterir o método , sendo a razão entre os fatores igual à:

#75
O primeiro passo é montar as reações que ocorrem no problema, perceba que o sal faz hidrólise.




O segundo passo é realizar os balanços de carga (BC) e de massa (BM).
BM: ![[Ba^{2+}] = Co(CO_3^{2-}) = [CO_3^{2-}] + [HCO_3^-] + [H_2CO_3]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_cbe7421759579ffbc05e96e433feb21b.gif?ssl=1)
BC: ![[H^+] + 2 \cdot [Ba^{2+}] = 2 \cdot [CO_3^{2-}] + [HCO_3^-] + [OH^-]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_2c8391a22d2695a8954eae78edcab372.gif?ssl=1)
Perceba que podemos deixar as equações referentes aos balanços de carga e massa em função de ![[H^+]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_7da3592729477229d8821cccd072f7b9.gif?ssl=1) e
e ![[CO_3^{2-}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_f591da2d14db506fbe928fed705b6631.gif?ssl=1) a partir das seguintes relações.
a partir das seguintes relações.
![K_{ps} = [Ba^{2+}][CO_3^{2-}] \Rightarrow [Ba^{2+}] = \frac{K_{ps}}{[CO_3^{2-}]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_54ed4e18f7a52abfed634858c7330646.gif?ssl=1)
![K_{a2} = \frac{[H^+][CO_3^{2-}]}{[HCO_3^-]} \Rightarrow [HCO_3^-] = \frac{[H^+][CO_3^{2-}]}{K_{a2}}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_3f2b0c061f4cbfa2071926a97fd81bf6.gif?ssl=1)
Somando as reações 2 e 3
![K_{a1} \cdot K_{a2} = \frac{[CO_3^{2-}][H^+]^2}{[H_2CO_3]} \Rightarrow [H_2CO_3] = \frac{[CO_3^{2-}][H^+]^2}{K_{a1}K_{a2}}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_7b516e22c323ab4e65e054fa8fc47bff.gif?ssl=1)
![K_w = [OH^-][H^+] \Rightarrow [OH^-] = \frac{K_w}{[H^+]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_4c0d923c13fa88cc857321122c2bd35a.gif?ssl=1)
Substituindo as novas relações nos balanços:
![BM: \ \ \frac{K_{ps}}{[CO_3^{2-}]} = [CO_3^{2-}] + \frac{[H^+][CO_3^{2-}]}{K_{a2}} + \frac{[CO_3^{2-}][H^+]^2}{K_{a1}K_{a2}}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_4952a78aeb667ec90cc5ff849447051a.gif?ssl=1)
![BC: \ \ [H^+] + 2 \cdot \frac{K_{ps}}{[CO_3^{2-}]} = 2 \cdot [CO_3^{2-}] + \frac{[H^+][CO_3^{2-}]}{K_{a2}} + \frac{K_w}{[H^+]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_fa67b02ef57fdec382f0b2fd9f9da678.gif?ssl=1)
Perceba que podemos colocar o termo em evidência na equação 1:
![\frac{K_{ps}}{[CO_3^{2-}]} = [CO_3^{2-}] \cdot \left(1 + \frac{[H^+]}{K_{a2}} + \frac{[H^+]^2}{K_{a1}K_{a2}}\right)](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_4a518400de79dfa0ec5f64d66a088fc6.gif?ssl=1)
Isolando o termo .
![[CO_3^{2-}] = \sqrt{\frac{K_{ps}}{\left(1 + \frac{[H^+]}{K_{a2}} + \frac{[H^+]^2}{K_{a1}K_{a2}}\right)}}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_960a9d178edd51bbd44b962b12559a61.gif?ssl=1)
Substituindo essa nova expressão na equação 2 temos:
![[H^+] + \frac{2 \cdot K_{ps}}{\sqrt{\frac{K_{ps}}{\left(1 + \frac{[H^+]}{K_{a2}} + \frac{[H^+]^2}{K_{a1}K_{a2}}\right)}}} = \sqrt{\frac{K_{ps}}{\left(1 + \frac{[H^+]}{K_{a2}} + \frac{[H^+]^2}{K_{a1}K_{a2}}\right)}}\cdot \left(2 + \frac{[H^+]}{K_{a2}}\right) + \frac{K_w}{[H^+]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_5a2162251240ff16b558a3bb7c6a3b3f.gif?ssl=1)
Perceba que agora temos uma equação com apenas uma variável, utilizando uma bela calculadora encontramos que ![[H^+] = 1,1 \cdot 10^{-10}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_ab912ba2df71ee6f3b9ecc03d70eeaaa.gif?ssl=1)
Agora podemos utilizar a fração das espécies para encontrar a solubilidade do 

![K_{ps} = S^2 \cdot \frac{K_{a1}\cdot K_{a2}}{[H^+]^2 + K_{a1}\cdot [H^+] + K_{a1} \cdot K_{a2}}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_00c61bdaa4a88b9feb009c8830968c52.gif?ssl=1)
Substituindo os valores dados e a concentração de íons hidrogênio na solução, encontramos que  .
.
#76
Em colisões orientadas são aquelas que estão na orientação espacial correta para que a reação ocorra, elas podem, ou não, ter energia o suficiente para que a reação ocorra. Por outro lado, as colisões efetivas são aquelas que, além de estarem orientadas, tem energia suficiente para que, ao ocorrer a colisão, a reação se concretize. Lembremos agora da equação de Arrhenius:

Neste caso, as colisões orientadas são representadas pela constante de Arrhenius ( ) e o termo
) e o termo  diz respeito a porcentagem de colisões orientadas que são efetivas, ou seja, que possuem energia o suficiente para que ocorra a reação.
diz respeito a porcentagem de colisões orientadas que são efetivas, ou seja, que possuem energia o suficiente para que ocorra a reação.
#77
A equação de Nerst é escrita da seguinte forma:

Sendo  e
e  e o quociente reacional
e o quociente reacional
![Q = \frac{[Produtos]}{[Reagentes]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_c694c7e45159234a2aee3933b55f3f9c.gif?ssl=1)
Então, a expressão será:
![E = E^0 - \frac{RT}{mF} \cdot ln\frac{[A^{m-}]}{[A]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_a5816118e508d054b0b6605b85ae4ffb.gif?ssl=1)
#78
Sabemos que ![E = E^0 - \frac{RT}{mF} \cdot ln(\frac{[A^{m-}]}{A})](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_23ac48ee2c631f11b51c983c3e198664.gif?ssl=1)
Além disso, também temos que
![A_0 = [A] + [A^{m-}] + [A(CO_2)_m^{m-}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_2dc9a7814dd2db29771bac1f40aa6a64.gif?ssl=1) (1)
(1)
![x_a' = \frac{[A^{m-}] + [A(CO_2)_m^{m-}]}{A_0}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_3f98e1d491f2fa34a14859441af82160.gif?ssl=1) (2)
(2)
![K = \frac{[A(CO_2)_m^{m-}]}{[A^{m-} \cdot P_{CO_2}^m]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_27163b4814e65061aee7638ad54f4b07.gif?ssl=1) (3)
(3)
Da equação (2), sabemos que ![x_a' \cdot A_0 = [A^{m-}] + [A(CO_2)_m^{m-}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_0ed0e7025abc0f643ef9e44df34b5737.gif?ssl=1) , substituindo em (1) temos:
, substituindo em (1) temos:
![A_0 = [A] + x_a' \cdot A_0 \Rightarrow [A] = A_0(1-x_a')](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_16146a46070aa5d91ad198b8f3d3d8a2.gif?ssl=1)
Podemos obter mais informações da equação (2):
![A_0 = ([A^{m-} + [A(CO_2)_m^{m-}])/x_a' \Rightarrow \frac{[A^{m-}]}{[A]} = \frac{x_a'}{1-x_a'} \cdot \frac{[A^{m-}]}{[A^{m-}]+[A(CO_2)_m^{m-}]}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_62966390e7d7bae4d9ba07472b7121ef.gif?ssl=1)
Veja que podemos simplificar o segundo termo da expressão dividindo tudo por ![[A^{m-}]](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_d7b0d8adca15623c2798af116fa9af33.gif?ssl=1) , assim:
, assim:
![\frac{[A^{m-}]}{[A]} =\frac{x_a'}{(1-x_a')} \cdot \frac{1}{1+\frac{[A(CO_2)_m^{m-}]}{[A^{m-}]}}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_3c97b5b1dfd8dde9dceb337216af4355.gif?ssl=1)
Mas da equação (3) nós temos que ![\frac{[A(CO_2)_m^{m-}]}{[A^{m-}]} = K \cdot P_{CO_2}^m](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_97c2be3ad6f2b297f4da7ac6be233bae.gif?ssl=1) , assim, vamos substituir o termo por
, assim, vamos substituir o termo por 
![\frac{[A^{m-}]}{[A]} =\frac{x_a'}{(1-x_a')} \cdot \frac{1}{1+K\cdot P_{CO_2}^m}](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_03d075f54833230f024a5cfc951b1075.gif?ssl=1)
Reescrevendo a equação em função do potencial:



Então
 e
e 
#79
A) Através da tabela presente na aula 2 de eletroquímica, podemos observar que as semi-reações que ocorrem na célula são as seguintes:
 ,
, 
 ,
, 
Com base nisso, podemos ver que a reação global da célula de Daniell é a seguinte:
 ,
, 
Utilizando a equação de Nernst, portanto, podemos observar como o potencial da pilha irá variar com a modificação das concentrações das espécies; da seguinte forma:

![E = 1,10 - \frac{0,059}{n}\cdot ln(Q) \rightarrow E = 1,10 - \frac{0,059}{2} \cdot ln(\frac{[Zn^{2+}]}{[Cu^{2+}]})](https://i0.wp.com/noic.com.br/wp-content/plugins/latex/cache/tex_c267aba2b6d460d92872703f4d9084a2.gif?ssl=1)

B) A energia pode ser entendida pelo da pilha nessa situação, que é expresso da seguinte forma:

Utilizando o mesmo esquema do item anterior, chegamos que o potencial nessa situação é:

Portanto, temos:

C) Veja que a ponte salina é o meio que faz com que a reação eletroquímica continue a sua duração, isto é, o meio no qual há a troca de íons e o consequente fluxo eletrônico. Por conta disso, a pilha não segue ocorrendo caso não haja a presença da ponte salina, fazendo com que o uso dela não seja verificado.
#80
A) É importante notar que a única grandeza que é conservada em um diagrama de Latimer é a energia livre das transformações. Por conta disso, para encontrarmos o potencial entre o  e o
e o  , deve-se realizar o seguinte:
, deve-se realizar o seguinte:




B) O diagrama de Frost resulta no seguinte:

#81
Para realizar esse item basta aplicar a fórmula da ebuloscopia, em que:  em que m é a molidade e i o fator de van't hoff. Devido a natureza do açúcar ser molecular, o i = 1, agora iremos calcular o resto. A molalidade da solução para esse caso é somente o número de mols de açucar dividido pela massa de água em Kg
em que m é a molidade e i o fator de van't hoff. Devido a natureza do açúcar ser molecular, o i = 1, agora iremos calcular o resto. A molalidade da solução para esse caso é somente o número de mols de açucar dividido pela massa de água em Kg
 , portanto,
, portanto,  é 0,015°C.
é 0,015°C.
#82
Seguindo o raciocínio do item passado, temos:  . Aqui temos a molalidade da solução multiplicada pelo fator de van't hoff, uma vez que os sais são completamente dissolvidos. Calculando, temos que = 0,33°C
. Aqui temos a molalidade da solução multiplicada pelo fator de van't hoff, uma vez que os sais são completamente dissolvidos. Calculando, temos que = 0,33°C
#83
Pela lei de raoult, temos que:  e que
e que  , pela lei de dalton temos que
, pela lei de dalton temos que  em que X representa a fração molar de A na fase liquída e Y na fase gasosa.
em que X representa a fração molar de A na fase liquída e Y na fase gasosa.
Tendo noção disso, podemos fazer o seguinte:  , já que a soma das frações molares é 1. Usando a lei de raoult de A e comparando com a lei de dalton, temos que
, já que a soma das frações molares é 1. Usando a lei de raoult de A e comparando com a lei de dalton, temos que  , logo,
, logo,  e isso pode ser rearranjado para
e isso pode ser rearranjado para  , então
, então  . Isolando
. Isolando  , temos a seguinte expressão:
, temos a seguinte expressão:  , invertendo os termos da equação obtemos a resposta:
, invertendo os termos da equação obtemos a resposta:  .
.
#84
![]()
![]() Podemos percerber que a equação desejada é somente uma manipulação das duas equações que temos os dados, então é
Podemos percerber que a equação desejada é somente uma manipulação das duas equações que temos os dados, então é  . Calculando, obtemos
. Calculando, obtemos 
#85
![]()
Podemos ver que essa equação desejada (3) só é mais uma manipulação das outras equações que já possuímos. Portanto,


#86
Assim como os outros itens, temos:
![]()
 .
.
= 
#87
Primeiramente vamos calcular a variação de temperatura pela propriedade coligativa da ebulioscopia:

Nesse sentido, devemos calcular a molalidade  e fator de Van'’ Hoff
e fator de Van'’ Hoff 
Vamos calcular primeiramente a molalidade

Nesse sentido, vamos calcular o número de mols do soluto por uma regra de três




Desse modo molalidade tem módulo de 
Depois disso devemos calcular o fator de van't hoff, que pode ser feito "quebrando" a molécula de  , obtem-se:
, obtem-se:
 e
e 
Percebe-se, portanto, que o fator de van't hoff é 
Agora, basta calcular a variação de temperatura pela fórmula

Agora, basta calcular a temperatura final pela fórmula:



Agora, respondendo à pergunta final: O ovo cozinhará mais rapidamente quando se adiciona sal à água, pois a temperatura de ebulição acaba por ser maior, logo, o ovo é capaz de absorver mais calor, cozinhando de forma mais eficiente.